Tercer Milenio
En colaboración con ITA
Genómica
Completando el mapa del genoma humano… 20 años después
En el mapa del genoma humano quedaban zonas sin secuenciar, regiones que hoy se exploran. ¿Por qué no han podido ser leídas hasta ahora?
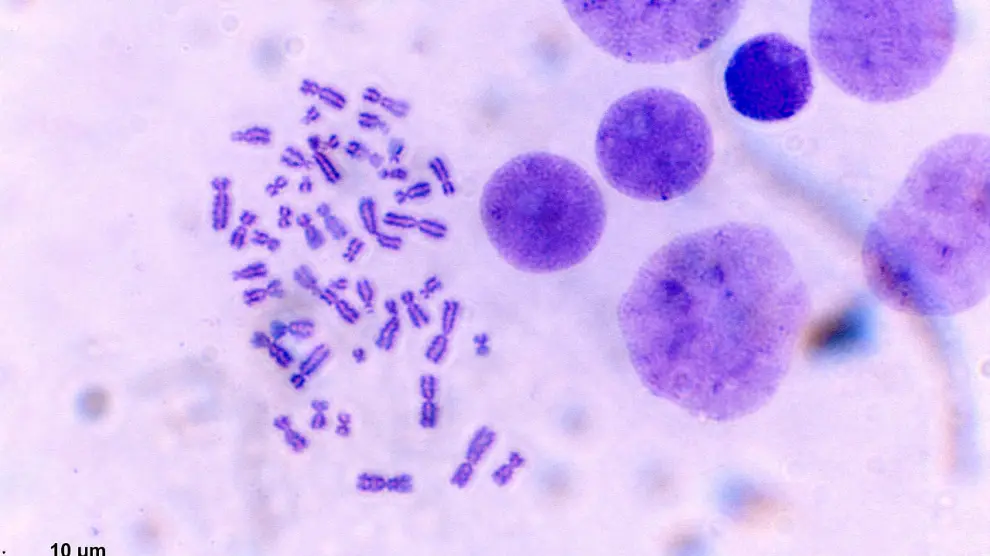
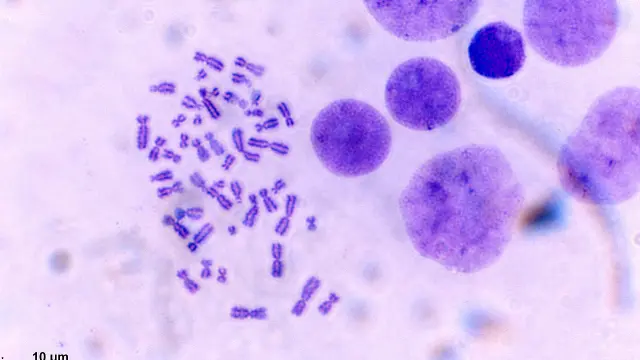
“Estamos aquí para celebrar la finalización del primer estudio de todo el genoma humano. Sin duda, este es el mapa más importante y maravilloso jamás producido por la humanidad”. Con estas palabras, el presidente de los Estados Unidos, Bill Clinton, daba inicio el 26 de junio del 2000 a la presentación del primer mapa del genoma humano. Clinton se dirigió al mundo desde el Salón Este de la Casa Blanca, acompañado por los embajadores del Reino Unido, Japón, Alemania, y Francia, y por el primer ministro británico Tony Blair, en conexión desde Londres vía satélite. También le acompañaban Francis Collins, director del Proyecto Internacional del Genoma Humano, y Craig Venter, presidente de Celera, la empresa que capitaneó la iniciativa privada.
En su discurso Clinton mencionó cómo, dos siglos antes, en ese mismo escenario, el presidente Thomas Jefferson y el capitán Meriwether Lewis habían contemplado otro mapa, resultado de otra expedición: la de este último y William Clark por el oeste americano, rumbo al Pacífico, para explorar y cartografiar el territorio de la Luisiana, recién adquirido por Estados Unidos en 1803. Según Clinton, el mapa de Lewis y Clark “fue un mapa que definió los contornos y expandió para siempre las fronteras de nuestro continente y nuestra imaginación”.
Uno y otro mapa tenían más elementos en común. El de Lewis y Clark estaba muy lejos de las actuales y precisas cartografías por satélite. Y el mapa del genoma humano, aún siendo uno de los hitos científicos del siglo presentado con todo el ceremonial en 2000, tampoco había logrado cartografiar completamente nuestro genoma. Tanto en el primer borrador como en las versiones posteriores, publicadas a lo largo de las dos últimas décadas, quedaba todavía cierto porcentaje de nuestro genoma sin secuenciar.
Un anuncio pendiente de publicación
El pasado 27 de mayo, un grupo internacional de científicos anunció haber secuenciado completamente por primera vez el genoma humano, incluso aquellas regiones ausentes en todas las versiones anteriores. El estudio, difundido inicialmente como una prepublicación, está pendiente de ser revisado y publicado en una revista científica.
¿Qué esconden estas regiones ignotas del genoma y por qué no han podido ser leídas hasta ahora? Karen Miga, investigadora de la Universidad de California en Santa Cruz, implicada en el proyecto, lleva años interesada en explorar este último territorio desconocido del genoma humano: “Nunca antes se había hecho y la razón por la que no se había hecho es porque es difícil”.
Viaje en el tiempo
Pero para entender el porqué de esta dificultad debemos viajar primero en el tiempo. El ambicioso Proyecto Genoma Humano se inició en 1990 y fue necesaria más de una década, la creación y colaboración de distintos institutos de secuenciación en Estados Unidos, el Reino Unido, Francia, Alemania, Japón y China, y 2.700 millones de dólares (unos 5.200 millones actuales), para completar el primer mapa del genoma humano. La secuencia del primer cromosoma, el número 22, se publicó en 1999. En junio de 2000, tuvo lugar la rueda de prensa en la Casa Blanca y, meses más tarde, el 15 de febrero de 2001, se publicaron dos artículos en las revistas ‘Nature’ y ‘Science’ en los que se presentaba y analizaba el primer borrador de la secuencia del genoma. Finalmente, en 2003, se declaró el proyecto como finalizado.
Para secuenciar (leer) un genoma entero, este debe romperse primero en fragmentos pequeños, ya que las máquinas que se emplean para secuenciar el ADN normalmente trabajan con fragmentos relativamente cortos. Una vez se dispone de las secuencias de los fragmentos, el genoma completo se ensambla como si se tratase de un puzle, uniendo en el ordenador las secuencias individuales aprovechando regiones de solapamiento que existen entre ellas.
En el caso del Proyecto Genoma Humano, los 3.000 millones de letras del genoma (3.000 Mb) se rompieron en fragmentos de 100 a 300 mil letras (kb) conocidos como cromosomas artificiales bacterianos o BAC, de los que se sabía su posición relativa en los cromosomas. Estos, a su vez, se fragmentaron en trozos más pequeños para poder ser secuenciados. El método de secuenciación que se empleó, denominado Sanger en honor a su inventor, el premio Nobel de Química, Frederick Sanger, permite leer fragmentos de unos pocos centenares de letras o bases de ADN (entre 800–1.000 con la actual metodología Sanger automatizada).
Este laborioso trabajo dio como resultado un genoma de referencia muy continuo y preciso, obtenido a partir de muestras de distintos voluntarios anónimos. No obstante, debido a las características de la metodología empleada, en la secuencia de referencia faltaba alrededor del 15% del genoma: amplias regiones constituidas por largas secuencias repetitivas, difíciles de incorporar en los BAC, de secuenciar, y mapear.
Aún así, la secuencia de referencia obtenida en el Proyecto Genoma Humano ha sido la guía que ha permitido el desarrollo de la medicina genómica y se ha empleado como patrón para poder ensamblar, con la ayuda de herramientas bioinformáticas, los centenares de miles de genomas individuales que se han ido secuenciando estos últimos años.
El boom de la genómica en la última década ha tenido lugar gracias al desarrollo de las tecnologías de secuenciación masiva del ADN (denominadas de última generación o NGS por sus siglas en inglés), que se basan en la secuenciación en paralelo de millones de fragmentos de entre 150 y 300 letras y en un ensamblaje posterior. Esta revolución, capitaneada por la empresa americana Illumina, ha reducido exponencialmente tanto el tiempo invertido en la secuenciación de un genoma como su coste.
El primer genoma tardó 13 años en completarse y actualmente se tarda unas pocas horas, y de centenares de millones de dólares se pasó, en 2014, al genoma de 1.000 dólares, y actualmente a unos 300.
También en estas últimas décadas se ha ido completando y corrigiendo la secuencia de referencia, trabajando en el 15% restante. No obstante en el último ensamblaje de 2013, seguía faltando un 8% del genoma. En esta fracción se encuentran regiones clave como las cercanas a los extremos de los cromosomas, los telómeros, y otras que constituyen los centrómeros y que contienen secuencias extremadamente repetitivas. Estos últimos han sido el foco de interés de Karen Miga, una de las investigadoras que ha liderado el reciente proyecto de secuenciación completa del genoma.
Los centrómeros pueden visualizarse en los cromosomas antes de la división celular, cuando nuestras células han duplicado su material genético y cada cromosoma pasa a tener dos cromátidas hermanas y su forma se asemeja a una aspa. El centrómero sería como una especie de nudo que mantiene juntas las dos cromátidas hermanas hasta el momento de la división celular para asegurar que las dos células resultantes heredarán el mismo número de cromosomas.
Desde que era estudiante, Miga se preguntaba cómo podía ser que unas estructuras tan esenciales para la vida, tan fundamentales para el funcionamiento de la célula, se encontrasen en regiones del genoma que eran como grandes mares de secuencias repetidas en tándem.
Por ello en 2019 creó, junto con Adam Phillippy, del National Human Genome Research Institute, y Evan Eichler, de la Universidad de Washington, el consorcio Telomere-to-Telomere (T2T), en el que ha participado una treintena de instituciones y que acaba de secuenciar por primera vez de forma completa los 3.055 millones de letras de ADN (o pares de bases) del genoma humano.
La secuencia de referencia que han obtenido los investigadores, denominada T2T-CHM13, contiene sin huecos las secuencias de los 22 autosomas (los cromosomas no sexuales) y del cromosoma X (uno de los dos cromosomas sexuales, que en las mujeres se encuentra en dos copias y en los hombres en una). También se han corregido numerosos errores, presentes en el genoma de referencia de 2013, y se han incorporado 200 millones de letras que corresponden a secuencias nuevas.
Entre las regiones que se han podido secuenciar por primera vez se encuentran los centrómeros de los cromosomas, y también 2.226 genes nuevos (copias de genes existentes) de los que 115 muy probablemente codifican proteínas y que incrementarían el total de genes humanos hasta 19.969.
Nuevas tecnologías de secuenciación
Este nuevo hito ha sido posible gracias a dos nuevas tecnologías de secuenciación, desarrolladas por la empresa californiana Pacific Biosciences y por la británica Oxford Nanopore, que permiten leer de una pieza largas moléculas de ADN sin necesidad de fragmentarlas.
La tecnología de Pacific Biosciences lee a tiempo real con detectores láser las señales que emiten las cuatro posibles letras de ADN (A, C, T, G) marcadas con moléculas fluorescentes, a medida que se van incorporando a la molécula de ADN en reacciones controladas que usan como molde el ADN que se quiere secuenciar. Con este método pueden secuenciarse moléculas de hasta 20.000 letras de ADN.
Por otro lado, la tecnología de Oxford Nanopore hace pasar la molécula de ADN a través de un nanoporo proteico (un pequeñísimo agujero) y monitoriza los cambios de corriente eléctrica que se producen. La señal resultante se decodifica para obtener la secuencia de letras del ADN.
Para la mayoría del genoma, los científicos han empleado la tecnología de Pacific Biosciences, mucho más precisa, y han utilizado la de Oxford Nanopore para completar algunas regiones.
El nuevo mapa del genoma obtenido por el proyecto T2T no procede de las células de una persona sino de una línea celular derivada de una mola hidatiforme (o embarazo molar), un crecimiento anormal en el útero de una mujer producido cuando un espermatozoide fertiliza un óvulo que no contiene núcleo. Generalmente las molas hidatiformes son diploides, y al igual que las células de una persona, contienen dos copias de cada uno de nuestros 23 cromosomas.
No obstante, estos son el resultado de la duplicación del material genético del espermatozoide y, por ello, los cromosomas de cada par son idénticos, incluidos los dos cromosomas X (46,XX). Los investigadores han optado por secuenciar estas células, porque su característica constitución cromosómica facilitaba enormemente las tareas computacionales necesarias para construir el mapa del genoma.
Aun siendo mucho más completa que la secuencia de referencia de 2013, el consorcio T2T estima que su genoma podría contener todavía un 0,3% de errores. Los investigadores tampoco han secuenciado el cromosoma Y, porque el esperma que originó la mola hidatiforme contenía un cromosoma X.
Regiones características de ciertos grupos humanos
El- T2T-CHM13 es el primer genoma completo obtenido con estas nuevas tecnologías de secuenciación. A partir de ahora se abre la puerta para secuenciar muchos más. Y esto es lo que pretende el consorcio T2T que se ha asociado con el Human Pangenome Reference Consortium para secuenciar en los próximos tres años los genomas de 300 personas de todo el mundo. De esta forma se podrán estudiar en profundidad regiones del genoma características (o incluso únicas) de distintos grupos humanos.
Por ejemplo, en 2018 un estudio identificó que los genomas africanos contienen regiones que no estaban presentes en el genoma de referencia. Estas comprenden 296 millones de pares de bases (un 10% más de ADN que los genomas no africanos). Contienen secuencias y genes nuevos, que procederían del genoma de poblaciones humanas 'fantasma' ya extinguidas pero que han dejado un rastro solo en algunos grupos humanos.
El genoma humano todavía esconde muchas sorpresas y secretos. Parafraseando el discurso de Clinton, hace 21 años, en la presentación del mapa del genoma y su mención al mapa de Lewis y Clark del oeste americano, podemos afirmar que la exploración del genoma en las últimas dos décadas ha expandido para siempre las fronteras de nuestra especie y nuestra imaginación.
-Ir al suplemento Tercer Milenio
Apúntate y recibe cada semana en tu correo la newsletter de Tercer Milenio