Tercer Milenio
En colaboración con ITA
Aquí hay ciencia
El genoma del coronavirus, la brújula para seguir su viaje por el mundo
Distintos países, entre ellos España, se encuentran en pleno proceso de desescalada de las estrictas medidas de confinamiento que se implantaron a mediados de marzo para hacer frente a la primera ola de la pandemia de covid-19. En la actual fase, el estudio del genoma de este nuevo coronavirus es una valiosa herramienta de vigilancia epidemiológica que permite a los científicos seguir en tiempo real la propagación del virus por el mundo. ¿Qué información podemos obtener del análisis del genoma del SARS-CoV-2?
En una pandemia como la actual, causada por un virus emergente, leer el genoma del virus puede darnos información sobre su origen, sobre cómo pasó de su reservorio animal a los humanos, y ayudar a los científicos a inferir cómo se ha propagado a lo largo y ancho del mundo. A escala local, puede emplearse para investigar si el virus ha entrado más de una vez en un país, cuál era su procedencia, cuántos brotes de covid-19 ha habido y cómo se relacionan entre sí.
A lo largo de la epidemia, un patógeno como el coronavirus SARS-CoV-2 acumula mutaciones en su genoma de forma aleatoria. En el interior de las células del huésped, el virus replica su genoma en un proceso de copia que es propenso al error. De forma análoga al juego del teléfono roto, los cambios introducidos en la secuencia del genoma se transmiten a la siguiente generación de virus y constituyen la materia prima para su evolución.
Comparando los genomas de virus obtenidos de centenares o miles de pacientes se puede analizar a qué velocidad muta el virus y si existen genes que incorporen más mutaciones que otros. También si alguna mutación puede tener un impacto en la virulencia o transmisibilidad del virus, aunque nada por ahora indique que esto haya ocurrido en el caso del coronavirus. Esta información puede ser muy útil para el desarrollo de fármacos antivirales y de vacunas, e incluso de métodos de diagnóstico molecular. Si las dianas moleculares de fármacos y vacunas mutan excesivamente estos podrían dejar de ser efectivos. Lo mismo pasa con los métodos diagnósticos, que se diseñan para detectar regiones muy conservadas del genoma; si estas mutan, su sensibilidad y especificidad se pueden ver afectadas.
En el caso del SARS-CoV-2, los datos parecen indicar que muta más lentamente que el virus de la gripe estacional. La base de datos GISAID ha estimado que incorpora 0,45 cambios por semana (menos de 25 mutaciones al año); por el contrario, la gripe estacional incorpora unas 50 mutaciones al año. Debido a que el genoma del SARS-CoV-2 es casi el doble de grande que el de la gripe, su tasa de mutación sería cuatro veces menor. Una buena noticia que puede ser clave para el desarrollo con éxito de vacunas, a diferencia de lo que ocurre en el caso de otros virus como el VIH.
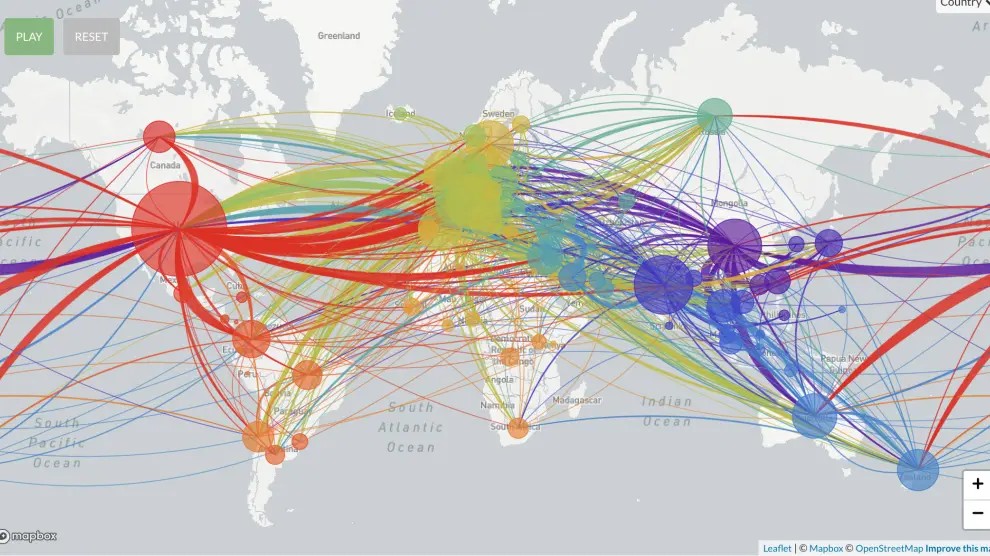
La comparación de miles de genomas de SARS-CoV-2 entre sí también sirve para analizar cómo evoluciona el virus en su viaje por los cinco continentes, pasando de persona a persona, e intentar anticiparnos a sus próximos movimientos. Las mutaciones en el genoma del virus nos indican cómo se relacionan entre sí los distintos virus secuenciados; actuarían de manera análoga a los sellos de un pasaporte, indicándonos en qué países ha estado el virus anteriormente.
El proyecto de acceso abierto Nextstrain, que entre otros patógenos rastrea en tiempo real la propagación y evolución de la gripe, el ébola o la tuberculosis, sigue también desde principios de año al SARS-CoV-2. Nextstrain emplea los datos genómicos públicos compartidos por investigadores de todo el mundo en GISAID y proporciona una visión de la epidemia que se actualiza constantemente, junto con potentes herramientas analíticas y de visualización de los datos que pone a disposición de la comunidad científica y médica. Todo ello con el objetivo de aumentar el conocimiento epidemiológico sobre la covid-19 y mejorar así las respuestas ante futuros brotes.
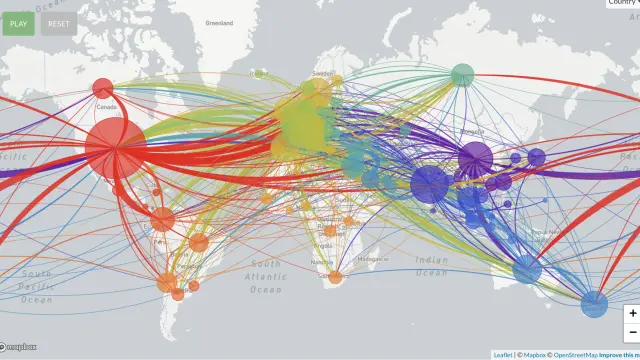
El proyecto Nextstrain permite ver la evolución de la pandemia. Aquí vemos un primer momento, centrado en Asia.
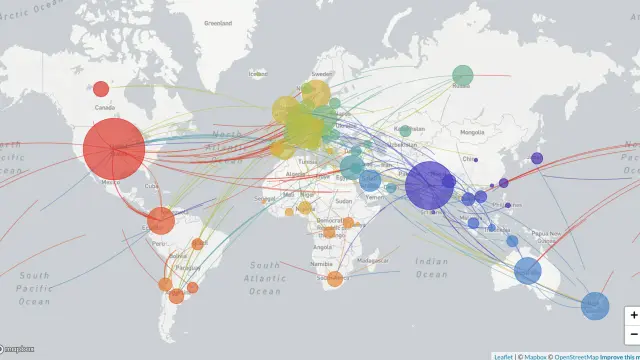
Plena pandemia: propagación global del SARS-CoV-2.
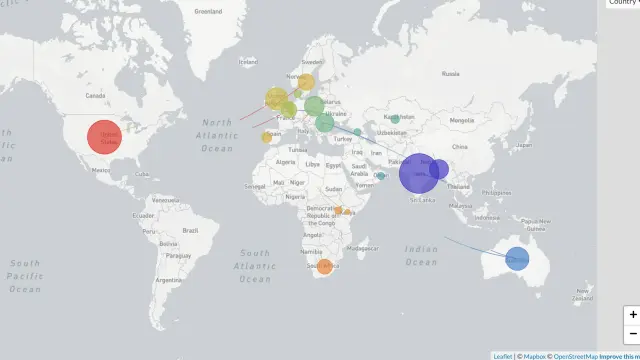
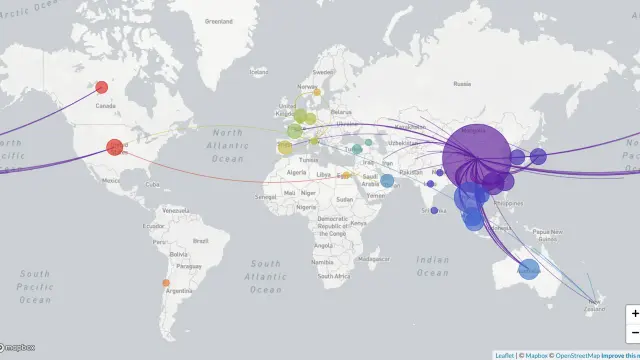
Visualización de los efectos del confinamiento de varios países, que logra que la mayoría de los focos ya no se relacionen.
Secuenciando genomas de SARS-CoV-2 para entender cómo funciona
Lo primero que nos viene a la cabeza cuando hablamos del genoma de un organismo es que contiene las claves para entender cómo funciona. En el caso de un virus, estudiando su genoma comprenderemos mejor qué estrategias sigue para infectarnos y multiplicarse, o qué factores son clave en su patogenicidad. Con esta información podremos desarrollar tratamientos eficaces para hacerle frente y vacunas para evitar contagiarnos.
En un hito sin precedentes, el primer genoma del SARS-CoV-2 se hizo público el 11 de enero, menos de dos semanas después de que China alertase a la OMS de un brote de neumonía de causa desconocida en la ciudad de Wuhan. Esta rapidez ha permitido, desde prácticamente el inicio de la epidemia, disponer de tests para el diagnóstico molecular de la covid-19, como los basados en la famosa técnica de PCR, que detectan en el paciente la presencia de material genético del virus.
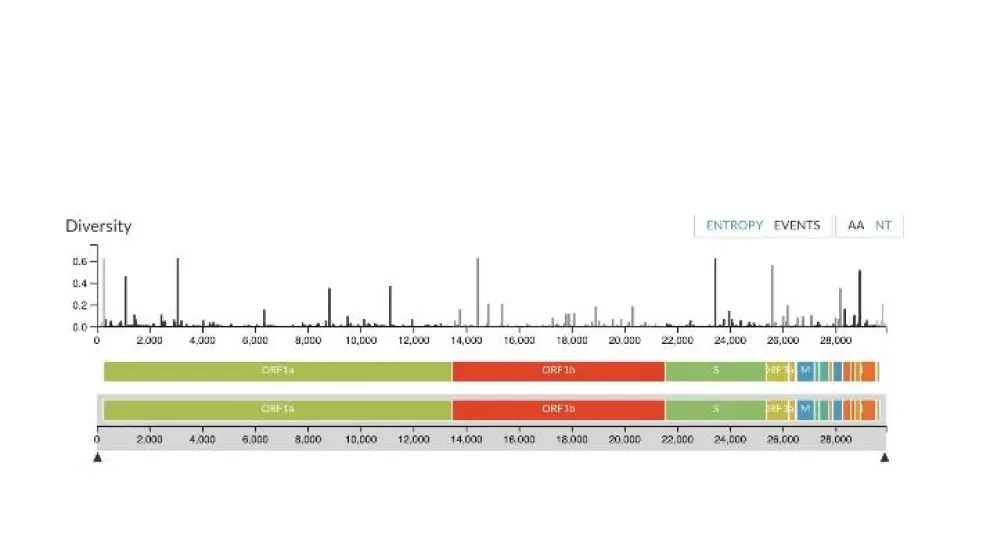
El SARS-CoV-2 es un betacoronavirus y su genoma está formado por un única cadena de ácido ribonucleico (ARN), una molécula prima hermana del ADN. La secuencia del genoma del SARS-CoV-2 está formada por un poco menos de 30.000 ‘letras’ de ARN y en ella se van repitiendo cuatro posibles letras (o bases): A (adenina), C (citosina), G (guanina) y U (uracilo); esta última sustituye a la T (timina) del ADN. Como referencia, es 100.000 veces más pequeño que nuestro genoma y sus genes codifican unas 29 proteínas, que son responsables, entre otras funciones, de la entrada del virus en las células, de la estructura de la cápside (el envoltorio exterior del virus), de la replicación de su genoma, de sabotear la maquinaria celular para forzar la fabricación de proteínas virales o de burlar las defensas tanto celulares como inmunológicas del huésped.

A partir de las muestras de un paciente, los investigadores generalmente extraen el material genético del virus, lo copian de ARN a ADN y, con unas máquinas denominadas secuenciadores de alto rendimiento, obtienen la secuencia completa del genoma: leen sus 30.000 letras. Actualmente laboratorios de todo el mundo siguen secuenciando genomas de SARS-CoV-2 y los comparten de forma abierta a través de la iniciativa GISAID (Global Initiative on Sharing All Influenza Data), una base de datos que nació hace 12 años con el objetivo de intercambiar información sobre el virus de la gripe. Hasta la fecha se han secuenciado más de 35.000 genomas de SARS-CoV-2. En España, las primeras secuencias de SARS-CoV-2 se obtuvieron a mediados de marzo.
El árbol filogenético de este virus crece desde Wuhan
Los biólogos moleculares emplean la información del genoma para construir árboles filogenéticos del virus, una suerte de árboles genealógicos, que les ayudan a rastrear las rutas que ha seguido en el espacio y el tiempo. Dos genomas virales con secuencias muy similares procederán de virus estrechamente emparentados que comparten un antepasado común reciente. No es que las mutaciones nos indiquen la procedencia del virus pero como cada muestra, cada genoma viral, procede de un paciente y está asociada al lugar del mundo donde se tomó, el árbol genealógico global permite rastrear las relaciones de los distintos genomas secuenciados y cartografiar el viaje del SARS-CoV-2 por el mundo.
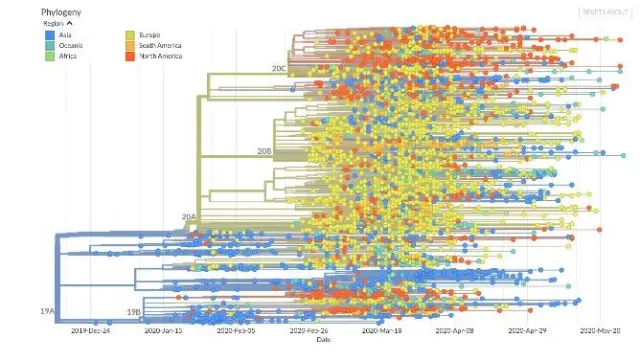
En esta representación de Nextstrain, cada punto representa un genoma y las ramas, las relaciones entre sí y cómo se han ido separando a lo largo del tiempo (en el eje horizontal), a medida que los distintos genomas del virus han ido incorporando mutaciones al azar. Se ve como en el inicio, solo hay virus de ‘procedencia asiática’ (en azul) y a partir de finales de enero van apareciendo y se diversifican los virus ‘europeos’ (en amarillo), norteamericanos (en rojo), etc.
Los datos de miles de genomas confirman que el coronavirus se originó en China, en Wuhan. A partir de la tasa de mutación del virus y de su árbol filogenético, los investigadores estiman que el salto desde su reservorio a los humanos se produjo en noviembre o a principios de diciembre de 2019. Si el virus hubiera estado dando vueltas en humanos sin ser detectado durante algún tiempo, los genomas de los virus de los primeros pacientes deberían tener un repertorio mayor de mutaciones. Aunque todavía se desconoce cuál es el reservorio natural del SARS-CoV-2, tiene similitud con coronavirus presentes en murciélagos, pero no se sabe si su transmisión a los humanos se produjo directamente o a través de otro animal. Los datos también indican que, por ahora, circula una única cepa del virus y que no existe una variante del SARS-CoV-2 que sea más virulenta, como publicaron erróneamente investigadores chinos a principios de marzo.
Cada punto representa un genoma y las ramas, las relaciones. Nextstrain
Epidemiología genómica
Proyectos de epidemiología genómica proliferan en todo el mundo. Nueva Zelanda o el estado de Victoria, en Australia, se propusieron desde el inicio de la epidemia secuenciar la mayoría de casos detectados.
- En Victoria (Australia) han secuenciado el 75% por ciento de los 1.700 casos de covid-19, lo que representa una de las iniciativas más exhaustivas de secuenciación de un brote infeccioso y planean usar los datos para detectar el origen de nuevos casos una vez se flexibilicen las restricciones en la circulación de personas.
- En Nueva Zelanda de momento han secuenciado el 25% del millar largo de casos del país, pero el objetivo es llegar al 70%. Los datos ya han servido para conectar casos que no habían sido relacionados entre sí mediante el rastreo de contactos y para detectar que lo que se creía era un solo brote de covid-19 en realidad eran dos brotes separados.
- En España, un estudio, coliderado por investigadores del Instituto de Biomedicina de Valencia (IBV-CSIC) y del Instituto de Biología Integrativa de Sistemas (I2SysBio-CSIC), comparará en una primera fase los genomas del coronavirus de 4.000 pacientes procedentes de 40 hospitales. Su objetivo en una segunda fase es llegar a los 20.000 genomas. El estudio busca entender el patrón de transmisión del virus en nuestro país, cómo llegó y en qué momento empezó a haber más transmisiones comunitarias que importaciones del virus desde el exterior. También si el hecho de que en un país existan zonas más afectadas que otras es debido a que se produjeron múltiples introducciones del virus en estas áreas o bien a otros factores como diferencias en la estructura de edades de la población. En España existe también un nodo de Next-Strain, Next-Spain, que recopila los datos obtenidos por distintos grupos de investigación españoles y los analiza para contribuir a la toma informada de decisiones sobre la evolución de la covid-19 en nuestro país.
-Ir al suplemento Tercer Milenio


.